
domingo, 26 de octubre de 2014
miércoles, 22 de octubre de 2014

lunes, 20 de octubre de 2014

Estimulación temprana
Es muy importante que nuestros niños con discapacidad y retos múltiples reciban estimulación temprana.aquí esta un manual muy práctico.

sábado, 18 de octubre de 2014
Síndromes
SÍNDROME DE APERT
Es una anomalía craneofacial hereditaria,
caracterizada por cierre prematuro de las suturas
del cráneo (craneosinostosis), aspecto
largo y puntiagudo de la cabeza (acrocefalia),
sindactilia (fusión de dedos en manos y pies) y
retrusión de tercio medio facial. Puede generar
presión intracraneal, problemas respiratorios,
oculares o de audición, etc.
Funcionalmente puede aparecer mayor, menor
o ausencia de retraso mental y alteraciones
neuropsicológicas (atención, lenguaje, función
ejecutiva…).
Es importante tener el diagnóstico médico.
A lo largo del proceso de evaluación e intervención,
como procesos continuos y circulares,
hay que tener en cuenta las ausencias escolares
de los afectados o afectadas por motivos
médicos (ingresos, operaciones, etc.).
La evaluación psicopedagógica debe cubrir
los siguientes aspectos: interacción del alumno
o alumna con el medio escolar, familiar y social,
aspectos emocionales, conductuales, neuropsicológicos
y pedagógicos. Todo desde una
perspectiva cualitativa y cuantitativa a través
de pruebas estandarizadas y/o observación,
entrevista a la familia, etc.
Si se valora la necesidad de una intervención
terapéutica extraescolar realizar la derivación
correspondiente: neuropsicólogo, neuropediatra,
logopeda, etc.

SÍNDROME DE CORNELIA DE LANGE
Es un trastorno congénito, presente al
nacimiento, no hereditario, caracterizado
por un conjunto de anomalías físicas, especialmente
faciales, y asociados a discapacidad
intelectual de grado variable. También
puede comprometer el crecimiento prenatal
y postnatal, y presentar malformaciones en
las extremidades.
Se da en 1 de cada 30.000 personas. Existe
un ligero predominio en mujeres.
Se hace por la constatación de signos clínicos,
principalmente faciales: cabeza pequeña,
pestañas largas, cejas espesas, labios finos y hacia
abajo, nariz pequeña y ancha. En el cuerpo
puede haber exceso de vellosidad, en las extremidades
presencia de anomalías y talla baja.
Presentan lenguaje alterado, por apraxia
oral-motriz y dificultades comprensivas, problemas
significativos en vista y oído, trastornos cardiacos
y gastrointestinales e hipersensibilidad.

SÍNDROME DE CRI DU CHAT O DE MAULLIDO DE GATO
El Síndrome Maullido de Gato o 5p-, es una
enfermedad cromosómica, congénita debido a
la falta de una parte del brazo corto del cromosoma
número 5. Se caracteriza por un llanto
distintivo que se asemeja al maullido de un gato
y que se va modificando con el tiempo.
Clínicamente se caracteriza por retraso
mental y del crecimiento. La enfermedad la
padecen una de cada cincuenta mil personas.
Las personas afectadas por esta anomalía, se
caracterizan por bajo peso al nacer, llanto característico
de tono alto similar al de un gato,
perímetro craneal reducido, cabeza pequeña
(microcefalia), ojos separados (hipertelorismo),
orejas de implantación baja. Las facies suelen
ser redondeadas, llenas y con frecuencia mofletudas
(cara de luna), paladar elevado y escarpado.
Presentan problemas en el habla y psicomotores,
estos últimos debido a la hipotonía.

SÍNDROME DE PRADER WILLI
El Síndrome de Prader-Wiili es una enfermedad
genética que afecta al cromosoma 15,
lo sufren uno de cada 10.000 nacidos.
Se presenta con hipotonía y dificultad de
alimentación en periodo neonatal, presentándose
en la infancia un apetito incontrolable
que conduce a la obesidad, retraso mental en
la mayoría de los casos y problemas conductuales.
Requieren de una atención multidisciplinar
(endocrinos, traumatólogos, logopedas,
fisioterapeutas, oftalmólogos, dentistas etc.)
Tienen necesidad constante de coger comida,
fragilidad emocional y problemas conductuales
que requieren un seguimiento, y como en todo
síndrome, cada afectado o afectada presenta
los síntomas con más o menos severidad.

SÍNDROME DE RETT
Es un trastorno neurológico que afecta principalmente
a niñas. La frecuencia de la enfermedad
es de 1: 15.000 niñas nacidas vivas. El
diagnóstico se basa en el cumplimiento de los
criterios de diagnóstico clínico. Éste se fundamenta
en la información de las primeras etapas
de crecimiento de la niña y del desarrollo y
evaluación continua de la historia médica y de
su estado físico y neurológico. El diagnóstico
clínico se puede confirmar con el estudio genético
de mutaciones en el gen MECP2.
El desarrollo de la niña es normal hasta los
6-18 meses de edad, momento en el que inician
una regresión.
Muchas de estas niñas caminan solas a la edad
que les corresponde, se sientan y comen con las
manos como cualquier otra niña, algunas pronuncian
palabras o frases, pero llega un momento en
que empiezan a perder estas capacidades.
Los síntomas que manifiesta la enfermedad
son; estereotipias en las manos, escoliosis,
apraxia, ataxia, ataques epilépticos, apneas, pies
pequeños y fríos, perímetro craneal reducido.

SÍNDROME DE X FRÁGIL
Es la forma hereditaria más común de discapacidad
intelectual, problemas de desarrollo,
comportamiento y aprendizaje. La causa de la
enfermedad es la falta de la proteína FMRP por
la mutación de un gen llamado FMR1, localizado
en el extremo del cromosoma X donde aparece
una fragilidad en los individuos afectados,
de ahí el nombre de X Frágil.
Los rasgos físicos son variables. A partir
de la pubertad se acompaña de dismorfia
(forma defectuosa de un aparato u órgano)
cráneo facial, con cara alargada, orejas grandes
y prominentes, macrogenitalismo (órganos
genitales externos agrandados), laxitud
articular, especialmente en los dedos de las
manos,... esta última es más frecuente que
se dé desde el nacimiento. Pueden presentar
además deficiencias visuales y auditivas, problemas
respiratorios, cardiacos y en algunos
casos, crisis epilépticas.
Es una patología hereditaria. Se estima que
1 de cada 2.500 hombres y 1 de cada 4.000
mujeres está afectado por el SXF.
Las características clínicas del Síndrome X
frágil abarcan una serie de conductas y de
rasgos físicos, más apreciables en niños que
en niñas. La afectación de la conducta incluye
hipotonía, retraso psicomotor, problemas
de aprendizaje, hiperactividad, falta de atención
y concentración, escasa comunicación
social, problemas de lenguaje y, en algunos
casos, comportamientos autodestructivos y
rasgos autistas por lo que pueden estar dentro
de los trastornos del desarrollo de etiología
desconocida, si no han sido diagnosticados
correctamente, que es el 80% de los
casos.
El diagnóstico del Síndrome X frágil se
realiza a través de una extracción de sangre
o de muestra de bulbo piloso, analizadas
utilizando técnicas de biología molecular.

Pilar Aguirre Barco, M.d. Manual de atención al alumnado con necesidades especificas de apoyo al alumnado con necesidades especificas de apoyo educativo por padecer enfermedades raras y crónicas. España: Junta de Andalucía
Epilepsia

La epilepsia tiene su origen en unos cambios
breves y repentinos del funcionamiento
del cerebro. Por esta razón, se trata de una
afección neurológica, la cual no es contagiosa
ni está causada por ninguna enfermedad o retraso
mental.
Las crisis epilépticas producen una alteración
momentánea del funcionamiento cerebral, debida
a la descarga súbita y desproporcionada
de los impulsos eléctricos que habitualmente
utilizan las células del cerebro. Esta descarga
puede afectar únicamente a una parte del cerebro
(crisis parciales o focales) o comprometer
a todo el cerebro (crisis generalizadas).
La epilepsia es un trastorno con muchas
causas posibles. Cualquier cosa que impida
o distorsione el patrón de actividad neuronal
normal puede conducir a la aparición de una
crisis epiléptica. Se ha observado que algunas
personas epilépticas tienen una cantidad más
alta de neurotransmisores activos (sustancias
encargadas de conducir el impulso nervioso
entre las neuronas), lo cual incrementa la actividad
neuronal. En otros se ha observado una
cantidad baja de inhibidores de dichos neurotransmisores,
lo cual también aumenta la actividad
neuronal. En ambos casos aparece la
epilepsia.
En países en vías de desarrollo la frecuencia
de padecer epilepsia es más alta debido al
pobre saneamiento ambiental que pone a la
población en riesgo de más enfermedades, a la
dificultad para la atención de los embarazos y
partos en zonas del campo alejadas de los servicios
médicos, etc. Alrededor del 75% de los
epilépticos inician el problema en las dos primeras
décadas de la vida, explicable porque el
cerebro inmaduro tiene más facilidad para producir
descargas anormales. La Sociedad Andaluza
de Neurología (SAN) afirma que alrededor
de 80.000 andaluces y andaluzas padecen esta
enfermedad neurológica.
Existen varias formas de presentación de las
crisis epilépticas. Las crisis generalizadas pueden
manifestarse con pérdida brusca de conocimiento
con caída al suelo, contractura de
los músculos de las extremidades y de la cara
seguidas de sacudidas rítmicas. En menores y
adolescentes, las crisis se presentan con una
pérdida de conocimiento, sin caída al suelo ni
convulsiones, de segundos de duración, con
rápida recuperación.
Las crisis parciales pueden presentarse con
sensaciones subjetivas extrañas o difíciles de
describir o con fenómenos auditivos, visuales,
sensación de hormigueo, etc. Estos síntomas pueden aparecer en forma aislada o dar paso
a una pérdida de conocimiento con movimientos
automáticos de la boca, de las manos o de
otra parte del cuerpo. En otras ocasiones las
crisis parciales pueden presentarse con sacudidas
de una extremidad o de la mitad de la cara,
sin pérdida de conocimiento.
Existen otras manifestaciones de crisis menos
frecuentes que deben ser evaluadas por el
especialista. Además, una persona puede presentar
más de un tipo de crisis.
Los distintos signos de alarma que advierten
que se está produciendo un ataque epiléptico
pueden ser los siguientes:
•• Períodos de confusión mental.
•• Comportamientos infantiles repentinos.
•• Movimientos como el de masticar alimentos
sin estar comiendo, o cerrar o
abrir los ojos continuamente.
•• Debilidad y sensación de fatigas profundas.
•• Períodos de “mente en blanco”, en los
que la persona es incapaz de responder
preguntas o mantener una conversación.
•• Convulsiones.
•• Fiebre.
La forma más típica, conocida también
como gran mal es con movimientos de las
4 extremidades, tipo sacudidas; se pierde la
conciencia y el control de los esfínteres, dura
segundos o algunos minutos, si persiste se
llama estado epiléptico. Otras formas son las
conocidas como ausencias frecuentes en niños
o niñas.
La posibilidad de controlar bien a un paciente
epiléptico, incluso por completo, es alta, de
un 80 – 85 % de los casos, e incluso, al cabo
de unos años, es posible la supresión completa
de la medicación.
La mayoría de los ataques epilépticos son
de breve duración y rara vez necesitan de asistencia
médica inmediata. Uno de los temores
de los profesionales de los centros educativos,
es que exista algún riesgo de perder la vida o
de sufrir un daño importante durante una crisis;
cuando esto rara vez ocurre.
¿QUE HACER EN CASO DE UNA CRISIS?
1. Permanecer calmado. Cuando se ha iniciado
la convulsión, usted no la podrá detener.
La mayoría de las convulsiones ceden espontáneamente
en menos de 3 minutos.
2. Despeje el área. Retire los objetos que
se encuentren cerca y puedan lastimar
al paciente, afloje la ropa apretada,
proteja su cabeza colocándole un objeto
suave debajo.
3. No introduzca ningún objeto en su
boca. Podrían lastimarlo o morderle los
dedos. No se ahogará con su lengua, ni
podrá evitar que se muerda.
4. Voltee la cabeza del paciente hacia
un lado. Y acuéstelo de lado para que
puedan salir las secreciones o vómito y
pueda respirar.
5. No se desespere. Si el paciente deja de
respirar o se le ponen morados sus labios,
es durante periodos breves y no
requiere de maniobras de resucitación,
ya que respirará nuevamente en forma
espontánea.
6. No se requiere de la presencia inmediata
de un médico o médica. Solamente
en caso que la convulsión tenga una
duración mayor de 5 a 10 minutos, o si
el paciente tiene una convulsión seguida
de otra.
7. Observe detalladamente qué es lo que
le ocurre. Es muy importante que visualice
como está ocurriendo la convulsión,
para así poder informar detalladamente
lo sucedido.
8. Sea amable con el niño o niña cuando
recupere la conciencia, manteniéndolo
calmado o calmada ya que puede avergonzarse.
9. No le ofrezca ningún alimento. Hasta
que esté totalmente alerta.
10. Permita que descanse o duerma. Ya
que les ayudará a recuperarse.
11. No suspender sus medicamentos anticonvulsivos.
12. Acudir al médico o médica. Después de
una convulsión, es necesario que sea
visto por su médico o médica para informarle
sobre la crisis.
Pilar Aguirre Barco, M.d. Manual de atención al alumnado con necesidades especificas de apoyo al alumnado con necesidades especificas de apoyo educativo por padecer enfermedades raras y crónicas. España: Junta de Andalucía
martes, 14 de octubre de 2014





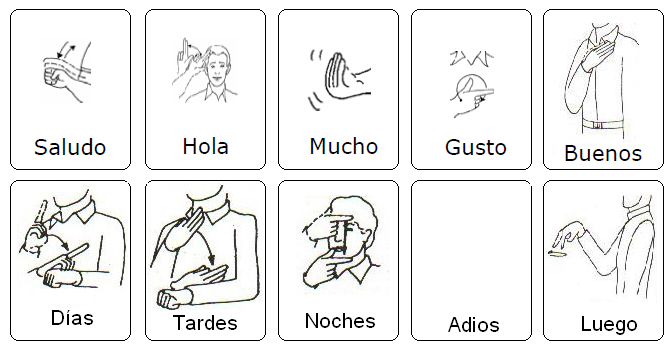
miércoles, 8 de octubre de 2014
.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)

.jpg)
.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
.jpg)

.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)

Suscribirse a:
Entradas (Atom)